Fastq¶
The alakazam package includes a set of functions to inspect the sequencing quality.
Example data¶
Load example data:
library(alakazam)
library(dplyr)
library(airr)
db <- read_rearrangement(system.file("extdata", "example_quality.tsv", package="alakazam"))
fastq_file <- system.file("extdata", "example_quality.fastq", package="alakazam")
Load quality scores¶
This method allows to add the quality scores to the repertoire data.frame as strings.
original_cols <- colnames(db)
db <- readFastqDb(db, fastq_file, style="both", quality_sequence=TRUE)
new_cols <- setdiff(colnames(db), original_cols)
db[,new_cols] %>% head()
## # A tibble: 1 × 4
## quality_num quality quality_alignment_num quality_alignment
## <chr> <chr> <chr> <chr>
## 1 90,90,90,90,90,90,90,90,90,90… {{{{{{… 90,90,90,90,90,90,90… {{{{{{{{{{{{{{{{…
The function readFastq takes as main inputs a repertoire data.frame (db) and
a path to the corresponding .fastq file (fastq_file). The sequencing quality scores will
be merged into the data.frame by sequence_id. The newly added columns are:
quality_num, quality, quality_alignment_num, quality_alignment. The other fields, contain the ASCII quality scores in the
form of a vector, where values are comma separated, and - or . positions
have value " " (blank).
After loading the quality scores with readFastqDb, getPositionQuality
can be used to generate a data.frame of sequencing quality values
per position.
quality <- getPositionQuality(db, sequence_id="sequence_id",
sequence="sequence_alignment",
quality_num="quality_alignment_num")
## Warning in FUN(X[[i]], ...): NAs introduced by coercion
head(quality)
## position quality_alignment_num sequence_id nt
## 1 1 90 CGCTTTTCGGATTGGAA C
## 2 2 90 CGCTTTTCGGATTGGAA A
## 3 3 90 CGCTTTTCGGATTGGAA G
## 4 4 90 CGCTTTTCGGATTGGAA C
## 5 5 90 CGCTTTTCGGATTGGAA T
## 6 6 90 CGCTTTTCGGATTGGAA G
min_pos <- min(quality$position)
max_pos <- max(quality$position)
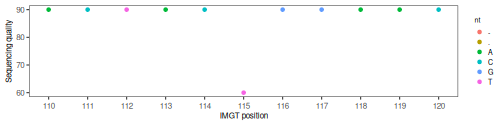
ggplot(quality, aes(x=position,
y=quality_alignment_num,
color=nt)) +
geom_point() +
coord_cartesian(xlim=c(110,120)) +
xlab("IMGT position") +
ylab("Sequencing quality") +
scale_fill_gradient(low = "light blue", high = "dark red") +
scale_x_continuous(breaks=c(min_pos:max_pos)) +
alakazam::baseTheme()
## Warning: Removed 27 rows containing missing values (`geom_point()`).
You can add use the quality data.frame to complement analysis performed
with other tools from the Immcantation framework. For example, you could inspect
the sequencing quality of novel polymorphisms identified with tigger, or
the sequencing quality in mutated/unmutated regions.
Mask low quality positions¶
Use maskPositionsByQuality to mask low quality positions. Positions with
a sequencing quality < min_quality will be replaced with an ‘N’. A message
will show the number of sequences in db that had at least one position
masked.
db <- maskPositionsByQuality(db, min_quality=70,
sequence="sequence_alignment",
quality="quality_alignment_num")
## Number of masked sequences: 1